
The shockwave of the pandemic accentuated the already long and laborious process of developing vaccines and therapeutics. As a result, the call for rapid design and testing of such products is evidently essential.
In the field of medicine development, there is an emerging tool that has potential to be used in a more rapid development process. These tools do not come from traditional lab experimentation, but from a computer science-based experimental space. Deep Machine Learning Models, in particular, are proving to be an advantageous stepping stone for developing
protein-based therapeutics for diseases.
To better understand deep learning models, we can examine our own process of learning. Deep learning models ‘learn’ to do tasks by example, just as a child might learn to behave in a particular way based on the actions of a parent. The model is tasked with performing classification of labeled data sets (photos, sounds, text, etc.). With this learning, the models are able to achieve extremely accurate assignments. They are trained using neural networks with many layers (Mathworks, (n.d.)). A neural network can be seen as an imitation of the way neurons in the brain work, where different neurons (cells in the brain) act like messengers and transmit information through signals to allow your body to process stimuli. Neurons are the translation of seeing an action and then physically reacting to it. This accuracy and speed are what are most needed in the design of therapeutics in response to new diseases/viruses.
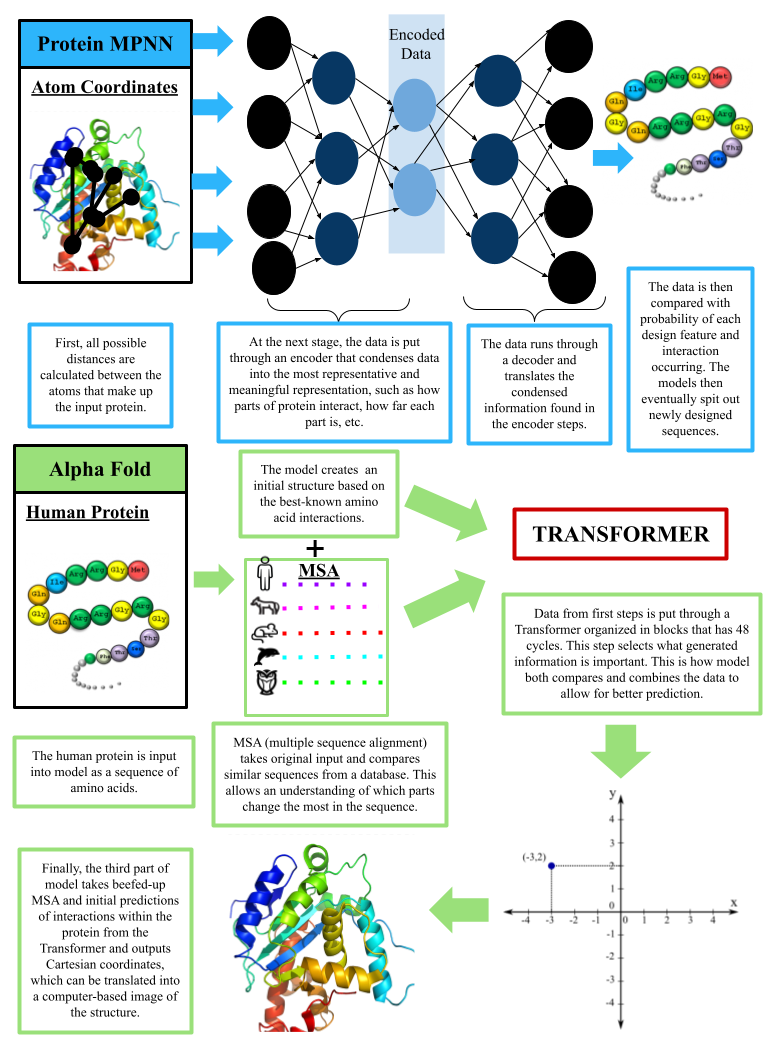
One of the biggest challenges in using proteins is determining how a protein will fold after it is made. Scientists still struggle to understand the various configurations of naturally occurring proteins, let alone synthetically designed proteins. In response to this knowledge gap, a program called AlphaFold is promising in predicting the folded states of proteins. AlphaFold was developed by DeepMind—Google’s AI company that looks to expand “general problem solving systems” (Deep Mind, (n.d.)). This deep learning model utilizes elements from similarly structured proteins in addition to past versions of the protein (Jumper et al., 2021) to identify similarities and create a structure of the predicted final and folded protein. Alpha fold has demonstrated extremely high accuracy ratings (David et al., 2022) and can be used to predict the folding of unknown natural proteins and, for the benefit of drug design, develop newly designed ones as well.
One of the focal groups that works to develop this software and learning model is David Baker’s lab at the University of Washington. Their most widely used work is Rosetta, which includes different packages that support different design goals (Leman et al., 2020). The Rosetta software is not a deep learning model, but rather a physics-based approach that uses well-known algorithms. In recent years, the Baker lab has released an even more useful protein design tool called Protein MPNN. Protein MPNN is a design algorithm that takes a base protein, redesigns said protein, and finally produces new sequences. Researchers who tested the designed proteins using MPNN found that the predicted sequence for the designed model had a recovery of 52.4%, compared with 32.9% in their other design program, Rosetta (Dauparas et al., 2022). Sequence recovery refers to how well the program was able to predict a protein sequence that would fulfill the intended role. Protein MPNN works by employing a series of layers of encoders and decoders that use statistics in addition to geometry to predict the best and most likely sequence that forms the protein in the real world. Encoders take information from input protein and translate it to a type of hidden or secret code. Only the decoders can interpret and translate it back into protein sequences that make sense. This system, with its high accuracy, is a game changer in the world of quickly designing proteins.
Although less highlighted in the mainstream media compared to popular AI tools such as ChatGPT, models like AlphaFold and Protein MPNN are game-changing. They provide scientists with greater opportunities to create tools for combating diseases and studying the intricacies of our biological world. New possibilities emerge with the assistance of AI, enabling speeds that would not be achievable by humans otherwise.
References
Alessia David, Suhail Islam, Evgeny Tankhilevich, Michael J.E. Sternberg, The AlphaFold Database of Protein Structures: A Biologist’s Guide, Journal of Molecular Biology, Volume 434, Issue 2, 2022, 167336, ISSN 0022-2836, https://doi.org/10.1016/j.jmb.2021.167336.
Deep Mind. (n.d.). Our Story. Retrieved from https://www.deepmind.com/about
Dauparas, J., Anishchenko, I., Bennett, N., Bai, H., Ragotte, R. J., Milles, L. F., Wicky, B. I. M., Courbet, A., de Haas, R. J., Bethel, N., Leung, P. J. Y., Huddy, T. F., Pellock, S., Tischer, D., Chan, F., Koepnick, B., Nguyen, H., Kang, A., Sankaran, B., Bera, A. K., … Baker, D. (2022). Robust deep learning-based protein sequence design using ProteinMPNN. Science (New York, N.Y.), 378(6615), 49–56. https://doi.org/10.1126/science.add2187
Jumper, J., Evans, R., Pritzel, A. et al. Highly accurate protein structure prediction with AlphaFold. Nature 596, 583–589 (2021). https://doi.org/10.1038/s41586-021-03819-2
Leman, Julia Koehler et al. “Macromolecular modeling and design in Rosetta: recent methods and frameworks.” Nature methods vol. 17,7 (2020): 665-680. doi:10.1038/s41592-020-0848-2
Mathworks. (n.d.). What Is Deep Learning?3 things you need to know. Retrieved from https://www.mathworks.com/discovery/deep-learning.html