
The human papilloma virus, or HPV for short, has been identified as the leading cause of cervical cancer and related tumors in women, with studies showing that approximately 10% of infected women will develop such conditions over the course of their lifetime. Although there are about 150 types of HPV, the most high-risk strains, “HPV-16 and HPV-18, are not only linked to cervical cancer but are also associated with gastrointestinal (anal) and head & neck (oropharyngeal) tumors,” (Pešut et al.). It’s also important to note that approximately 80% of HPV-associated cervical tumors are caused by the HPV-16 strain, while about 20% are attributed to HPV-18. Yet, these strains have only recently received extensive scientific attention within the past few decades. In order to better prevent the development of cervical cancer, understanding the biochemical and biophysical mechanisms of HPV’s viral replication cycle, as well as the complex interactions between the virus and host cells that ultimately drive tumor formation, is crucial.
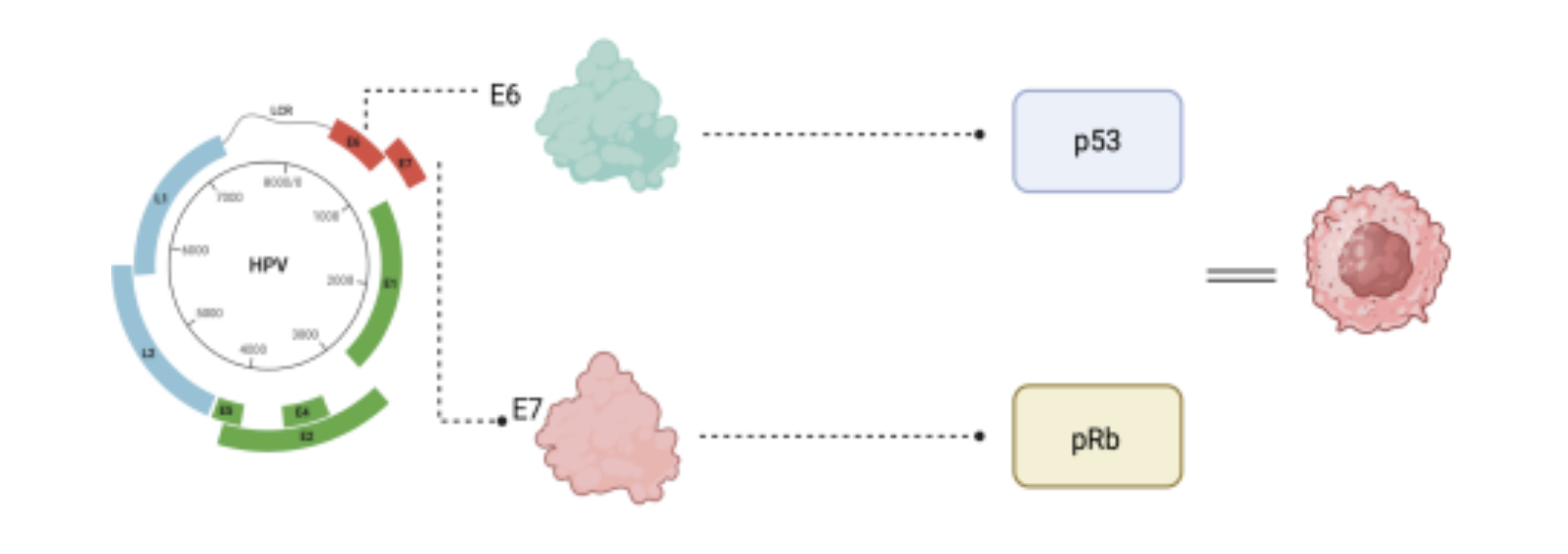
Oncoproteins are proteins that disrupt normal cell regulation and contribute to the development of cancer. In the case of HPV, recent discoveries have shown that “two viral oncoproteins, E6 and E7, along with the E5 protein, are the primary drivers of HPV-associated complications,” (Yim & Park). Although the role of the E5 protein is not entirely understood by researchers, it has shown to play a role in the differentiation dependent stages of the HPV life cycle, which leads to unexpected DNA production in normally inactive skin cells, enabling the virus to amplify its genome using the host’s DNA replication machinery. Studies also show that in mice, “the E5 protein, when induced in conjunction with E7, leads to a more aggressive progression of cervical cancer,” (Tomaić) . This cooperation is a common pattern in carcinogenesis and unfortunately, often results in the development of tumors. Therefore, the two most important viral oncoproteins cooperate extensively as previously mentioned; E7 drives early carcinogenesis whilst E6 accelerates progression towards malignancy.
The E6 protein from high-risk HPV strains is crucial in the development of cervical cancer because it disables several important defenses that normally protect cells from becoming cancerous. Its most well-known action is “breaking down a key tumor-suppressor protein called p53, using another protein called E6-AP,”(Yim & Park). This prevents damaged cells from undergoing apoptosis (a type of programmed cell death) or prevents them from repairing their DNA. E6 also blocks p53 in other ways, like stopping it from entering the cell’s nucleus or being chemically activated. Beyond p53, “E6 interferes with other cell death pathways by destroying proteins such as BAK, FADD, and caspase-8, and also disrupts a receptor called TNFR1”, which normally all help trigger cell death when needed (Garnett et.al). Therefore, the E6 protein drives a cycle of uncontrolled cell growth and survival through such processes. E6 also activates an enzyme called telomerase (hTERT), which gives infected cells the ability to divide indefinitely, which normal cells cannot do.
In contrast, HPV’s E7 protein also plays a major role in cervical cancer by causing cells to multiply uncontrollably and preventing them from maturing into their proper forms. Primarily, the protein is recognized for its “ability to break down proteins like retinoblastoma proteins (pRb), which usually act as brakes on cell division,” (Tomaić). When these pRb proteins are removed, proteins called “E2F transcription factors are released, pushing the cell to start copying its DNA in preparation for division,” (Tomaić). E7 also increases the activity of proteins that drive the cell cycle, like CDK2, while suppressing inhibitors (such as p21 and p27), further removing the cell’s internal checks on growth. Beyond controlling growth, E7” prevents skin cells from properly developing by degrading a regulatory protein called PTPN14 that is involved in cell function and signaling; it additionally interferes with transforming growth factor beta (TGF-B),” (White et.al.). E7 also helps the virus “evade detection by the immune system by shutting down key immune pathways (like cGAS-STING and NFκB) and silencing immune genes through chemical modifications to DNA,” (White et.al.). It overall promotes genetic instability by causing stress during DNA replication, activating DNA damage responses, and disrupting chromosome organization, leading to genetic errors.
Therefore, the disruptions caused by the E6 and E7 proteins, both independently and in collaboration, play a critical role in promoting the degradation of tumor suppressors, the inhibition of cell death, and ultimately, the progression of HPV-associated cancers. Understanding these mechanisms not only aids scientific progression, but also brings us closer to developing treatments that could prevent the HPV infection altogether. By recognizing how these proteins drive cancer development, researchers have a better chance at finding real solutions to a disease that continues to affect millions of women worldwide.
 References:
References:
- Tomaić, Vjekoslav. “Functional Roles of E6 and E7 Oncoproteins in HPV-Induced Malignancies at Diverse Anatomical Sites.” Cancersvol. 8,10 95. 19 Oct. 2016.
- Yim, Eun-Kyoung, and Jong-Sup Park. “The role of HPV E6 and E7 oncoproteins in HPV-associated cervical carcinogenesis.” Cancer research and treatment vol. 37,6 (2005): 319-24.
- Pešut, Ena et al. “Human Papillomaviruses-Associated Cancers: An Update of Current Knowledge.” Viruses vol. 13,11 2234. 6 Nov. 2021.
- Garnett, T O et al. “Accelerated degradation of FADD and procaspase 8 in cells expressing human papilloma virus 16 E6 impairs TRAIL-mediated apoptosis.” Cell death and differentiation vol. 13,11 (2006): 1915-26.
- White, Elizabeth A et al. “High-Risk Human Papillomavirus E7 Proteins Target PTPN14 for Degradation.” mBio vol. 7,5 e01530-16. 20 Sep. 2016.